The Growing Use of the FDA Breakthrough Devices Program and Recalls
At the close of 2025, the US Government Accountability Office (GAO) issued a report concluding that the Food and Drug Administration (FDA) should dedicate more resources to its oversight of recalls for medical devices.[1] If the use of a medical device begins to prompt safety or other concerns, either the FDA can mandate a recall or—more commonly—the device’s manufacturer can voluntarily initiate a recall and work with the FDA to resolve those concerns.[2] Yet, the GAO concluded the FDA does not have enough staff to oversee these recall processes, and even suggested policymakers consider whether the agency needs more authority to conduct recalls for devices.
The GAO report, however, does not touch specifically on the FDA’s newer Breakthrough Devices Program.[3] This Program has operated for about a decade and provides device manufacturers with a menu of potential benefits—including potentially modifying clinical trial expectations—if the FDA determines that a device they are still developing meets certain statutory criteria. To be designated a breakthrough, a device should “provide for more effective treatment” of serious conditions and either 1) use “breakthrough technologies,” 2) have no alternative on the market, 3) be better than alternatives on the market, or 4) generally be “in the best interest of patients.”[4]
In a recent article in the Columbia Science and Technology Law Review, I argue that the kind of expedited regulatory review that happens in the Breakthrough Devices Program should not be combined with immunity from tort liability.[5] Medical devices that are authorized through a Premarket Approval by the FDA are shielded from much state-level tort liability by federal law, which appears to be true even if those devices participate in the Breakthrough Devices Program and receive expedited regulatory review. This is a problem, since it could shift some of the risks of medical innovation onto patients who use new devices, instead of the companies that develop them. The argument was based in part on empirical findings illustrating how this Program has been operating over its first decade.
In light of the recent GAO report on device recalls, this blog post draws out and highlights three of the article’s findings that have potential relevance to medical device recall law and policy. They are:
- The number of breakthrough designated devices that go on to receive FDA authorization is increasing over time;
- A notable amount of these authorized breakthrough devices are “cleared” through the 510(k) pathway, which typically does not require clinical trials proving safety and effectiveness; and
- Recalls for breakthrough devices have already begun for this relatively new Program, affecting about one in ten of those authorized so far.
Authorizations of Breakthrough Devices Are Increasing
Generally, new medical devices (except for many low-risk ones) must get some kind of authorization from the FDA before they can be legally marketed in the United States. The three most common kinds of authorization are Premarket Approval, 510(k) clearance, and the De Novo pathway.[5] Premarket Approval is typically for higher-risk devices and requires clinical trials showing they are safe and effective. Moderate-risk devices instead usually use the 510(k) or De Novo pathways. The 510(k) process often does not require clinical trials if a manufacturer can show their device is “substantially equivalent” to a device already on the market. The De Novo process is for devices that are not equivalent to an existing product but are not risky enough to merit a full Premarket Approval, and may include some clinical trials.
Devices destined for any of these three kinds of authorization are eligible to apply to the Breakthrough Devices Program. The FDA reports how many medical devices it annually designates as “breakthroughs” on its website, which is roughly increasing over time.[3] However, the agency does not publicize which devices or manufacturers have received the designation or what regulatory benefits they received, if any, citing its own regulations and Freedom of Information Act exceptions for “confidential” business information.[6] In fact, the FDA did not even begin reporting which breakthrough devices had been authorized to enter the US market for patient use at all until 2022, when journalists began questioning the agency’s lack of transparency.[7]
Cross-referencing that now-public list of authorized breakthrough devices against other publicly available FDA databases gave a more robust picture of the Breakthrough Devices Program. The findings from my study show that the number of breakthrough devices that the FDA has authorized—not just designated—is also increasing over time.[5]
FDA Device Authorizations After Breakthrough Designation
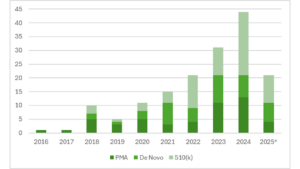
*Data are through June 30, 2025; reproduced with permission from the Columbia Science and Technology Law Review
More Breakthrough Devices Going Through the 510(k)
These data on authorized breakthrough devices also show that a surprising number of devices going through the Breakthrough Devices Program end up being authorized through the 510(k) pathway. In the last full year of data that was available at the time, over half of all authorized breakthrough devices that year (2024) went through the 510(k) process.
This finding is surprising, since for that to happen a breakthrough 510(k) device would seemingly need to both be a “breakthrough” but, at the same time, be “substantially equivalent” to an existing product. One of the requirements for using the 510(k) pathway is that the device either has “the same technological characteristics” as an existing device, or has “different technological characteristics” but “[d]oes not raise different questions of safety and effectiveness.”[8]
Since the FDA rarely reports which statutory criteria justified providing breakthrough designation to a device intended for the 510(k) pathway,[5] it is difficult to determine exactly how a “breakthrough” device could either use existing technology or use new technology that has no new safety or effectiveness implications. Perhaps these devices received the designation by meeting statutory criteria other than the use of “breakthrough technologies,”[4] though it is difficult to know based on only publicly available records.
Recalls for Breakthrough Devices Have Already Begun
The data also show that, as of mid-last year, one breakthrough device had already undergone a Class I recall and 16 devices underwent Class II recalls—involving 17 out of the 160 total breakthrough devices that had been authorized by then. Class I recalls happen when there is “a reasonable probability that [a device] will cause serious adverse health consequences or death,” while a Class II recall generally indicates a risk of “temporary or medically reversible adverse health consequences.”[9] For context, the GAO found that device manufacturers voluntarily initiated between 736 and 865 recalls each year, from 2020 to 2024.[1]
While the one Class I recall was for a device that underwent Premarket Approval, the Class II recalls were for breakthrough devices authorized across all three pathways. These recall data do not necessarily speak to the overall safety of the affected devices, as recalls can happen for a variety of reasons and can be remedied.
Yet, the notable percentage of breakthrough devices being recalled (over one in ten) may deserve greater policy attention. As further context, previous research has found that medical devices that receive priority review from the FDA—a benefit also included in the Breakthrough Devices Program—are generally recalled earlier and more often than devices receiving standard review.[10]
Implementing the GAO Recommendations
When read in light of the GAO report and previous research, these data suggest that the FDA may need even greater staffing and resources to monitor breakthrough device recalls, possibly over and above the boost already recommended by the GAO. These innovative devices may undergo less strict regulatory review as a part of their participation in the Breakthrough Devices Program,[6] which may then require a higher level of regulatory surveillance and recall oversight after patients begin using those devices to ensure that public health can be adequately protected.
While the current political moment may not facilitate providing the FDA with adequate resources for regulatory supervision, policymakers should seriously consider how to implement the GAO’s recommendations as early as possible, and may need to account for breakthrough devices when they do.
References
[1] U.S. Gov’t Accountability Off., Medical Device Recalls: HHS and FDA Should Address Limitations in Oversight of Recall Process, GAO-26-107619 (Dec. 8, 2025), https://www.gao.gov/products/gao-26-107619.
[2] See 21 C.F.R. §§ 7.40–7.59, 810.1–810.17 (2026).
[3] U.S. Food & Drug Admin., Breakthrough Devices Program, https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program (accessed May 13, 2026).
[4] 21 U.S.C. § 360e–3(b) (2026).
[5] Walter G. Johnson, Breakthrough or Breakaway Innovation?, 27 Colum. Sci. & Tech. L. Rev. 171 (2026), https://doi.org/10.52214/stlr.v27i1.14548.
[6] U.S. Food & Drug Admin., Breakthrough Devices Program: Guidance for Industry and Food and Drug Administration Staff (2023), https://www.fda.gov/media/162413/download.
[7] Katie Palmer & Mario Aguilar, FDA’s Breakthrough Device Program, Meant to Benefit Patients, Is Delivering the Biggest Gains for Companies, Stat News (2022), https://www.statnews.com/2022/04/18/fda-breakthrough-device-designation-investigation/.
[8] 21 C.F.R. § 807.100(b)(2) (2026).
[9] 21 C.F.R. § 7.3(m) (2026).
[10] Caroline Ong, Vy K. Ly & Rita F. Redberg, Comparison of Priority vs Standard US Food and Drug Administration Premarket Approval Review for High-Risk Medical Devices, 180 JAMA Internal Med. 801 (2020).